Biological macromolecules fold already during their biosynthesis either into stable three-dimensional (3D) structures or into multiple interconverting states to populate an ensemble of so-called intrinsically disordered states. Computational structural biology has the purpose to develop models that leverage the knowledge of the 3D structure of macromolecules to investigate the sequence-structure-function paradigm and shed light into the atomic-level mechanism of function.
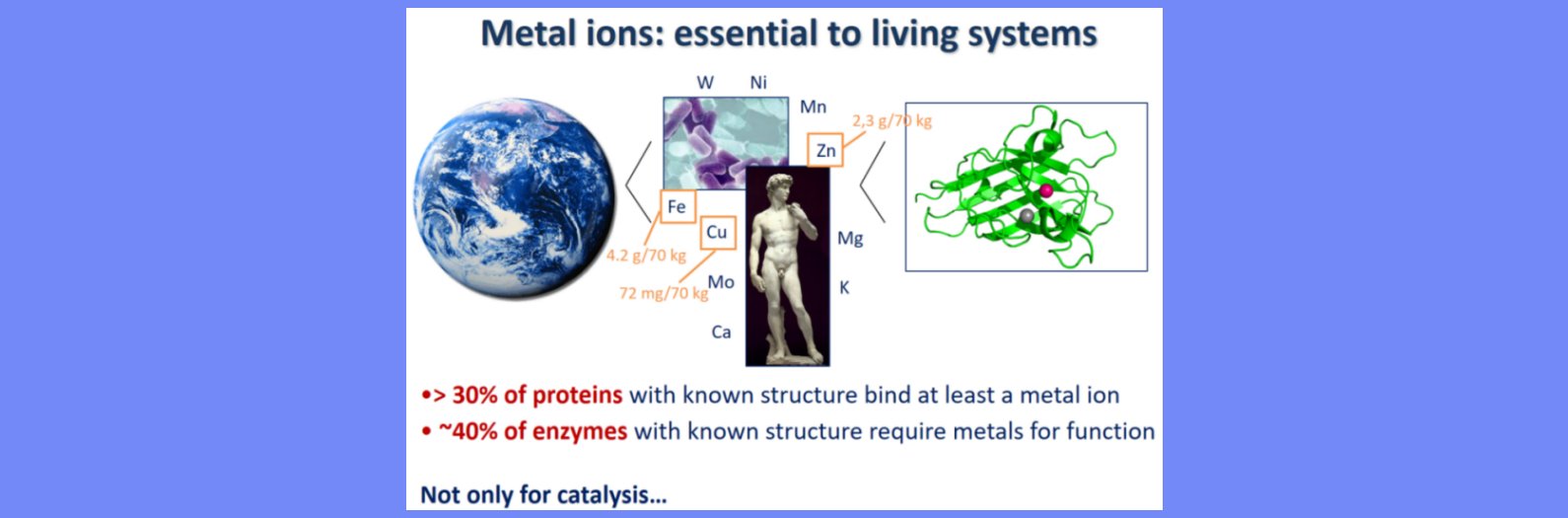
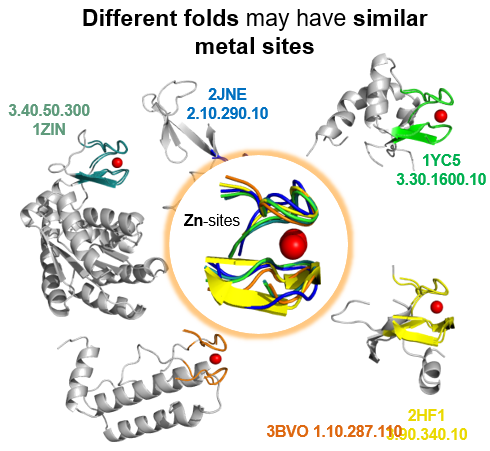 Computational structural biology is a broad discipline that encompasses many different application types. Over the years the focus of CERM researchers has been on metalloproteins, by a combination of the following methods: sequence-based prediction of 3D structure, mainly to investigate the variability of metalloprotein structure throughout evolution as well as the impact of mutations on the reactivity of metal-binding sites; modeling of protein-protein interactions, in order to understand how the individual components of macromolecular adducts recognize each other, what is the role of metal ions in such recognition processes and experimental NMR data can be best exploited to improve the modeling of adducts; archiving and classifying metal-binding sites, based on their specific structural features and regardless of the overall macromolecular fold with the aim of uncovering structural relationships that become apparent only at the local level; prediction and comparative analysis of metalloproteomes at the whole organism level, in order to characterize the influence of an organism’s lifestyle on its usage of metal ions.
Computational structural biology is a broad discipline that encompasses many different application types. Over the years the focus of CERM researchers has been on metalloproteins, by a combination of the following methods: sequence-based prediction of 3D structure, mainly to investigate the variability of metalloprotein structure throughout evolution as well as the impact of mutations on the reactivity of metal-binding sites; modeling of protein-protein interactions, in order to understand how the individual components of macromolecular adducts recognize each other, what is the role of metal ions in such recognition processes and experimental NMR data can be best exploited to improve the modeling of adducts; archiving and classifying metal-binding sites, based on their specific structural features and regardless of the overall macromolecular fold with the aim of uncovering structural relationships that become apparent only at the local level; prediction and comparative analysis of metalloproteomes at the whole organism level, in order to characterize the influence of an organism’s lifestyle on its usage of metal ions.